
EFI - Enzyme Similarity Tool
EFI-EST and Cytoscape Tutorials
SSN input -- General considerations
EFI-EST uses the UniProtKB protein sequence database (maintained by EMBL-EBI) for its annotations because, it provides the ability for members of the community to modify and/or correct functional annotations. In addition, EFI-EST uses the Pfam and InterPro databases (also maintained by EMBL-EBI) to provide easy access to the complete memberships of a large number of curated protein families/superfamilies (16,712 families for Pfam 31.0; 30,876 families/domains/sites for InterPro 64.0). The InterPro database collects signature sequences from 12 different databases, including Pfam, to define its families. Because the different databases may define the "same" family with slightly different signature sequences, InterPro families almost always are larger than Pfam families.
The sequence similarity networks generated by this webserver utilize the full length sequences of the proteins that are identified via their UniProt accession IDs (by BLAST in Option A, members of specified Pfam and/or InterPro families in Option B, the headers in a FASTA file in Option C, when read, and from lists of accession IDs in Option D). As a result, the clusters that are generated and visualized in the networks will result from sequence similarities for the entire sequence.
Many proteins have multiple domains; for these proteins the alignments used to calculate the alignment scores will not necessarily be for the domain in which you may be interested. However, we provide an "Advanced Option" for Option B that provides the capability to trim the full length sequences of multidomain proteins to generate SSNs using domain boundaries defined by Pfam for the Pfam family that you enter. We recommend that you use this advanced option carefully—Pfam families "always" contain fragments of full-length sequences plus domains often are interrupted by insertions, both potentially complicating the interpretation of the SSN.
INPUT: Four options for generating SSNs are available.
Select the option you want to use and enter the required information. For each input method, an "Advanced Options" menu allowing modification of the default parameters is available.
By default, the all-by-all BLAST used to calculate the edges for the SSN returns a result only if the e-value is ≤ 10-5.
We recommend SSNs to be generated with the 10-5 default value and an examination of the percent identity quartile plot to determine whether the default value should be changed. For short sequences, e.g., < 100 residues, this e-value may be too small to allow an alignment score corresponding to 30% or less to be used for filtering in the Analyze Data step. The "Advanced options" menu for each option allows to select a larger upper limit for the e-value by entering an integer ≤ 5 (the negative log of the e value); the lower limit for the input is 0.
After the input has been entered for any of the four options on the start screen, as shown in Figure 1, enter your e-mail address (for data retrieval only; blue arrow), and hit "Submit Analysis" at the bottom of the screen (green arrow). EFI-EST will assemble the sequence dataset and perform the all-by-all BLAST. The all-by-all BLAST will return alignment scores/edges for those sequence pairs for which the BLAST e-values are less than an upper limit threshold of 10-5 (or a different threshold specified in the 'Advanced Options'). For most families, the default threshold should provide sufficient internode connections (edges) in the networks that inferences about divergent evolution of protein function are possible.
If you are interested in detailed exploration of sequence-function relationships in families with more than 150,000 sequences, please submit a summary of your interests via the feedback form at http://efi.igb.illinois.edu//feedback.php and we may be able to assist.

Figure 1. Entire EFI-EST starting page.
Option A: Single sequence query
Networks for close homologs to a user-supplied sequence. Paste a protein sequence (without a FASTA header) into the input box (red arrow). A sequence dataset will be built containing the most closely related sequences retrieved from the UniProtKB database using a BLAST e value upper limit threshold of 10-5. A default of 1,000sequences is used, but the dataset may be smaller if < 1,000 sequences are found using a BLAST alignment score upper limit of 10-5. A default of ≤ 1,000 sequences is used because, in most cases, a full network with all sequences (nodes) will be viewable without having to collapse nodes into representative nodes (explained here). Use this option if you are only interested in those proteins that are most similar to your protein of interest.
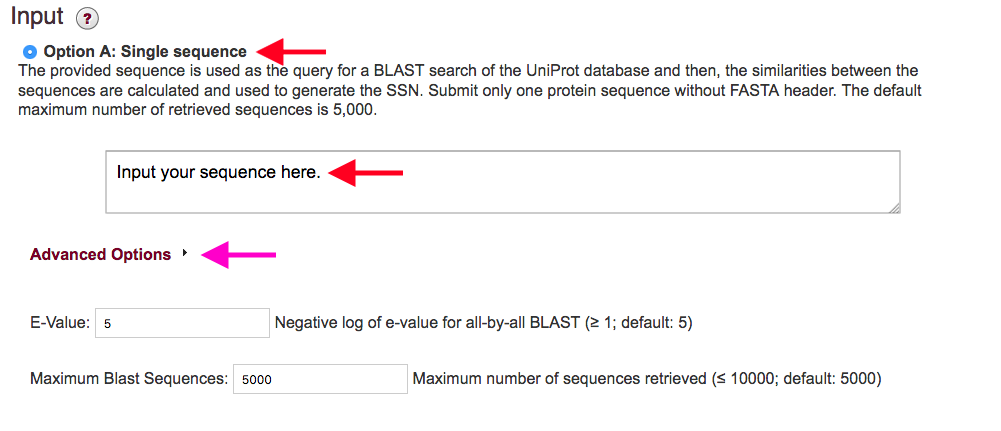
Figure 2. Settings for Option A.
Advanced Options (magenta arrows): By clicking on the Advanced Options tab below the input box, you can enter "custom" values for the maximum number of sequences that will be collected and the e-value used.
Maximum BLAST Sequences: Option A allows the user to collect a subset of sequences. It is possible to collect a maximum of 10,000 sequences. This option may be preferred if a full family network is difficult to handle in Cytoscape on memory limited computers. Alternatively, you can download a representative node network to visualize larger networks.
Option B: Pfam and/or InterPro families
Defined protein families are used to generate the SSN.
Pfam and/or InterPro family identifier(s) for your family of interest are used as input. The Pfam and/or InterPro families to which proteins belong to can be determined on the Pfam and InterPro websites.
More than one Pfam and/or InterPro family number(s) can be entered as the input for Option B, in a comma-separated list (red arrow). The number of sequences that can be used in Option B is limited to ≤150,000. This limit is set to ensure that assembling the dataset/performing the all-by all BLAST as well as generating the networks for most families can be completed within several hours (very large families may require several days). When the dataset is complete, you will receive an e-mail with a link to analyze the dataset. This link will be active for 40 days so that you may return at your convenience.
When an entry is recognized, the sequence count per family and estimated total count (there may be redundancy between families) are displayed (blue box).
Option B usually will result in a much larger dataset than Option A because all of the members of families are included. Full networks may be problematic to open in Cytoscape on memory limited computers when large families are analyzed. As an alternative to full networks, representative node networks are available to download on the result page.
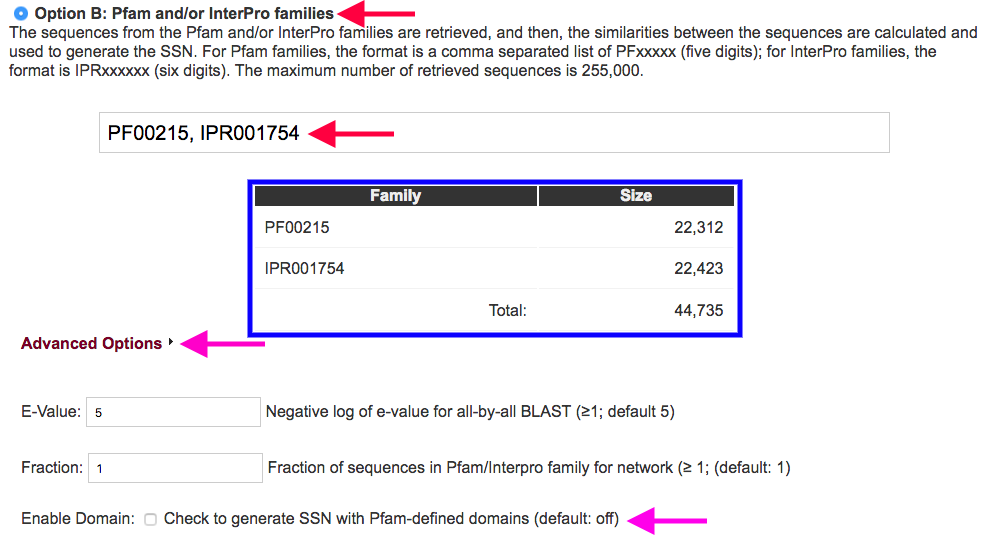
Figure 3. Settings for Option B.
Advanced Options (magenta arrows): By clicking on the "Advanced Options" menu, you can enter a "custom" e-value used in the all-by-all BLAST. You also can select a fraction of the sequences in the input Pfam and/or InterPro family(ies) so that you can generate an "overview" of the families you are interested in. You can also choose to generate the SSN with the Pfam defined domains instead of the full-length sequences.
Fraction: If the dataset you initially select is too large (> 150000 sequences) you can select the same dataset and specify a fraction of that dataset to be analyzed. This decreases the number of sequences, but provides representative overview of the original dataset. The value entered represents the divisor by which you wish to fractionate the dataset, e.g., 10 = only every 10th sequence in the total sequence dataset is used. The Uniprot sequence dataset is not preorganized, so the sampling is "random".
Domains: It is difficult/impossible to infer the functional relationships between proteins that possess a single domain and ones composed of multiple domains using SSNs. Pfam defines N- and C-terminal domain boundaries for members of its families based on sequence, not structure, comparisons. Using these domain definitions, it is possible to trim full-length sequences of multi-domain proteins to obtain only the domain specified by the Pfam family ID.
For example, in nonribosomal peptide synthases (NRPSs), the domain definitions can be used to extract the individual domains (e.g, condensation domains, PF00668) and use these to generate a SSN. If the full-length sequence has multiple homologues of the same domain, all of the domains will be extracted and used to generate the SSN.
By using the "Enable Domain" option, the SSN will be generated with the sequences from the defined domain instead of the full-length sequences. In the networks, the N- and C-boundaries of the domain are appended to the UniProt accession ID for the full-length sequence (ID:N-terminus:C-terminus). This makes the produced SSN incompatible with the generation of a corresponding GNN and the use of the coloring utility.
Please be aware that Pfam families "always" include at least some fragments of full-length sequences as the result of sequencing errors, so these may complicate the analyses of networks for domain. In addition, in some proteins the domain belonging to one family may be inserted in the domain for a second family, resulting in two pieces of the second domain in the network.
Option C: User-supplied FASTA file
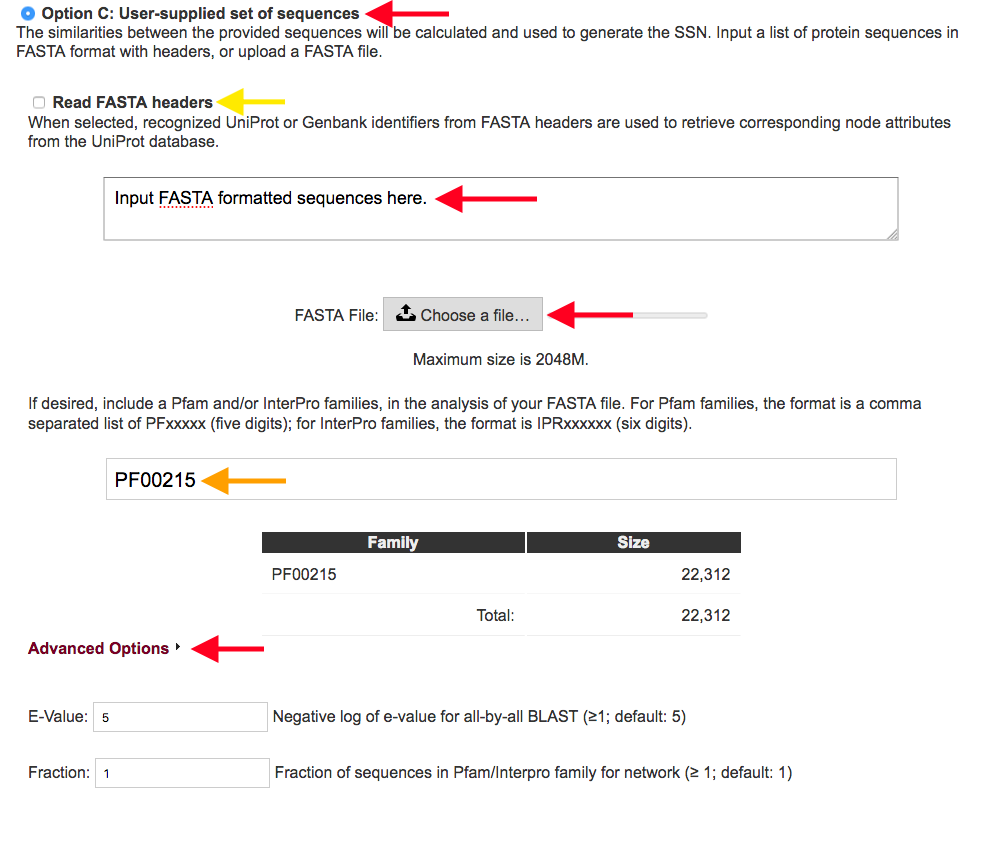
Figure 4. Settings for Option C.
Option C allows the user to input protein sequences in a FASTA format, using the direct input box or by uploading a file, and generate an SSN using those sequences (red arrows). The sequences submitted can be enriched with sequences from specified Pfam and/or InterPro families so that the provided sequences can be placed in the context of a protein family (orange arrow). When a protein family is supplied for enriching your initial submission, the number of sequences from this family is displayed, for information.
Option C provides two further options for handling the FASTA file (yellow arrow).
By default, the sequences from the FASTA file are used for generating the SSN. All characters of the FASTA header are used as the “Description” node attribute in the SSN for the corresponding protein sequence, and the number of residues is the value of the “Sequence_Length” node attribute. In addition, “shared name” and “name” node attributes are assigned individually to each sequence, and numbered sequentially starting with 0. The preceding characters (to make 6) in the “shared name” and “name” node attributes will be "z", e.g., zzz123.
If the option is activated by ticking the box, the FASTA header for each sequence is read, to import the accession IDs. UniProt IDs and/or NCBI IDs (RefSeq IDs, UniProt/Swiss-Prot IDs, GenBank IDs, PDB IDs, and/or “retired” NCBI GI numbers) present in the FASTA header are identified (following the formatting “rules” described below). A UniProt ID is used to directly identify the sequences and annotations for SSN node attributes in the UniProt database. An NCBI ID is used to query the idmapping file provided by UniProt to identify the equivalent UniProt ID, and the sequence and annotations for SSN node attributes are obtained from the UniProt database. For these entries (with UniProt or NCBI IDs in the header), two additional node attributes will be present: “Query_IDs” will list the UniProt and/or NCBI ID(s) from the FASTA header and “Sequence_Source” will indicate “USER”.
Not all NCBI IDs will identify an equivalent UniProt ID (the NCBI database is larger than the UniProt database). For these entries, the default information (FASTA header as the Description and Sequence Length) will be provided.
If the user enters Pfam and/or InterPro families IDs (orange arrow), the node attributes associated with these sequences will include “FAMILY” as the “Sequence_Source” node attribute. If a node is associated with both the FASTA file and a sequence from Pfam/InterPro family, the “Sequence_Source” node attribute will be “FAMILY+USER”.
The NCBI BLAST server provides FASTA files in which multiple FASTA headers often are provided for the same sequence. As a result, more than one header/accession ID may identify the same UniProt ID. Also, files from the NCBI BLAST can contain entries for the PDB structures of mutant proteins: the PDB ID for a mutant often will identify the UniProt ID for the wild type protein, so multiple PDB entries (for the wild type and mutant proteins) will identify the same UniProt ID. When this occurs, the SSN will contain a single node for the UniProt ID, and the “Query ID” node attribute will contain a list of all of the NCBI IDs that located the UniProt ID.
If a UniProt ID cannot be located for a sequence in the UniProt format because it is more recent than our database or the NCBI ID cannot be located in the idmapping file, the default information (FASTA header as the Description and Sequence Length) will be provided.
Two outcomes are possible if an NCBI ID cannot be located in the idmapping file:
- If the FASTA header is one of several associated with the same sequence (files from the NCBI BLAST server) and a UniProt ID can be identified for at least one of the headers, the NCBI ID will be included in the “Other_IDs” node attribute for each of the UniProt IDs that are identified for the sequence. The “shared name” and “name” attributes will have “z” format described previously.
- Otherwise, the sequence in the FASTA entry will be used for the SSN. As described for Option C, the “shared name” and “name” node attributes have a total of six characters. The sequences in the FASTA file are numbered sequentially starting with 0. The preceding characters (to make 6) will be "z", e.g., zzz123. The NCBI ID is included in the “Other_IDs” node attribute. If the sequence has more than one FASTA header with an NCBI ID that cannot be retrieved, all will be included in the “Other_IDs” node attribute.
When the “Read FASTA headers” is not selected, the FASTA header is not interrogated for an accession ID and is used only as the “Description” node attribute. The sequence in the FASTA file is used to generate the SSN. The node “name” and “shared name” node attributes will be generated as described two paragraphs above, e.g., zzz123. The sequences from the FASTA file will have USER as the “Sequence_Source”.
The acceptable formats for FASTA headers are provided in the following examples taken from output files from the UniProt and NCBI BLAST servers (accession ID highlighted):
UniProt (from UniProt BLAST; TrEMBL and SwissProt, respectively)
>tr|R9RJF1|R9RJF1_PSEAI Mandelate racemase OS=Pseudomonas aeruginosa PE=4 SV=1
>sp|P11444|MANR_PSEPU Mandelate racemase OS=Pseudomonas putida GN=mdlA PE=1 SV=1
NCBI RefSeq (from NCBI BLAST)
>WP_016501748.1 mandelate racemase [Pseudomonas putida]
NCBI UniProt/Swiss-Prot ID (from NCBI BLAST)
>Q0TE80.1 RecName: Full=Enolase; AltName: Full=2-phospho-D-glycerate hydro-lyase; AltName: Full=2-phosphoglycerate dehydratase
NCBI GenBank ID (from NCBI BLAST)
>AAA25887.1 mandelate racemase (EC 5.1.2.2) [Pseudomonas putida]
NCBI PDB ID (from NCBI BLAST)
>pdb|1MDR|A Chain A, The Role Of Lysine 166 In The Mechanism Of Mandelate Racemase From Pseudomonas Putida: Mechanistic And Crystallographic Evidence For Stereospecific Alkylation By (r)-alpha-phenylglycidate
NCBI GI Number (from NCBI BLAST; now retired)
>gi|347012980| 4-O-methyl-glucuronoyl methylesterase [Myceliophthora thermophila ATCC 42464]
Option C also accepts FASTA headers in which the IDs (formats described in Option D) immediately follow the “>” symbol, e.g., the following headers abbreviated from those shown above:
UniProt
>R9RJF1
>P11444
NCBI RefSeq
>WP_016501748.1
NCBI UniProt/Swiss-Prot ID)
>Q0TE80.1
NCBI GenBank ID
>AAA25887.1
NCBI PDB ID
>1MDR
NCBI GI Number (now retired)
>347012980
Advanced Options (magenta arrows): By clicking on the Advanced Options tab below the input box, you can enter a “custom” value used in the all-by-all BLAST. You also can select a fraction of the sequences in the input Pfam and/or InterPro family(ies) so that you can generate a “representative” network for families ≤ 150,000 sequences.
Fraction: This advanced option applies ONLY to the sequences in the Pfam or InterPro family if so specified, not in the user-supplied FASTA file. As in Option B, although the limit on the number of sequences that can be used to generate a SSN is limited to ≤ 150,000, with this advanced option you can select a fraction of the total number of sequences for larger sequence sets to generate a network.
Option D: SSNs for a user-supplied text file of accession IDs.

Figure 5. Settings for Option D.
The user uploads a text file containing UniProt IDs, NCBI IDs (RefSeq IDs, UniProt/Swiss-Prot IDs, GenBank IDs, and/or “retired” GI numbers), and/or PDB IDs (red arrows). These are the most commonly encountered sequence database accession IDs that users may have for their “favorite” proteins.
A UniProt ID is used to directly identify the sequences and annotations for SSN node attributes in the UniProt database. An NCBI ID is used to query the idmapping file provided by UniProt to identify the equivalent UniProt ID, and the sequence and annotations for SSN node attributes are obtained from the UniProt database. For these entries (with UniProt or NCBI IDs in the header), two additional node attributes will be present: “Query_IDs” will list the UniProt and/or NCBI ID(s) from the FASTA header and “Sequence_Source” will indicate “USER”.
The formats for UniProt IDs, NCBI IDs, and PDB IDs are described below with examples:
UniProt IDs
UniProtKB ID is 6 or 10 alphanumerical characters in the following formats:
1 2 3 4 5 6 7 8 9 10
[O,P,Q] [0-9] [A-Z,0-9] [A-Z,0-9] [A-Z,0-9] [0-9]
[A-N,R-Z] [0-9] [A-Z] [A-Z,0-9] [A-Z,0-9] [0-9]
[A-N,R-Z] [0-9] [A-Z] [A-Z,0-9] [A-Z,0-9] [0-9] [A-Z] [A-Z,0-9] [A-Z,0-9] [0-9]
For example:
P11444
T2HDW6
A0A0A7PVN6
NCBI RefSeq IDs
An NCBI RefSeq ID is 2 letters followed by an underscore followed by a series
of digits, a period, and one or more digits for the sequence version number,
e.g.,
WP_016501748.1
NP_708575.1
YP_002409124.1
NCBI UniProt/Swiss-Prot IDs
An NCBI UniProt/Swiss-Prot ID is the UniProt ID followed by a period and one or
more digits for the sequence version number, e.g.,
Q31XL1.1
B7LEJ8.1
C4ZZT2.1
NCBI GenBank IDs
The format for NCBI GenBank IDs is 3 letters followed by five digits, a period,
and one or more digits for the sequence version number, e.g.,
BAN56663.1
AAC15504.1
BAM38409.1
PDB IDs
The format for PDB IDs is one digit followed by two letters and a digit/letter:
1MDL
1MRA
3UXL
NCBI GI Numbers
An NCBI GI number (now retired) is a series of digits.
Sequences and annotations may not be retrievable for NCBI IDs, PDB IDs, and GI numbers because “equivalent” UniProt matches could not be located in the UniProt idmapping file (the UniProt database is smaller than the NCBI database; some GI numbers may not be currect).
Option D reads the accession in the user-uploaded text file. For a UniProt ID, the sequence and annotation information is retrieved IDs from our local database downloaded from UniProt. Some UniProt IDs may not be in the database used to generate SSNs—because our database is downloaded with every other release of the UniProt database (every 8 weeks), the user’s input file may contain more recent UniProt IDs that are not in our database.
When an NCBI ID. PDB ID, or GI number is located in the idmapping file provided by UniProt, the “equivalent” UniProt ID is used to retrieve the sequence and annotation information from our database. In the SSN, the identity of the NCBI ID, PDB ID and/or GI number is included in the “Query_ID” node attribute.
Not all NCBI IDs and GI numbers are included in the idmapping file because the UniProt database is smaller than the NCBI database, so sequences and annotations will not be retrieved for some of the NCBI IDs. For these IDs, the ID is added to the “nomatch” list that can be downloaded from the “Analyze Data” page. In the nomatch file, UniProt IDs that could not be located are designated “NOT_FOUND_DATABASE"; NCBI and PDB IDs that could not be located are designated “NOT_FOUND_IDMAPPING”. When several IDs are locating the same Uniprot IDs, DUPLICATE is mentioned within the source attribute column information.
The SSNs generated with Option D provide a node attribute (“Query ID”) that associates the UniProt IDs in the SSN (in the “name” and “shared name” node attributes) with the NCBI IDs, PDB IDs, and GI numbers provided in the input file. Multiple NCBI and PDB IDs can be associated with the same UniProt ID; if/when this occurs, the node attribute is a list of the IDs associated with the UniProt ID. This node attribute can be searched in Cytoscape so that the user can locate the sequences/node attributes for the input accession IDs.
As described for Option C, the user can specify one or more Pfam and/or InterPro families to be included in the SSN. The node attributes for the sequences in the Pfam/InterPro family members will be those provided in Option B. The SSN includes a node attribute that specifies whether the sequence is associated with a sequence in the input file (USER) or Pfam/InterPro family (FAMILY).
Advanced Options: same as those described for Option C.
Advanced Options (magenta arrows): By clicking on the Advanced Options tab below the input box, you can enter a “custom” value used in the all-by-all BLAST. You also can select a fraction of the sequences in the input Pfam and/or InterPro family(ies) so that you can generate an “overview” network for families ≤ 150,000 sequences.
Fraction: This advanced option applies ONLY to the sequences in the Pfam or InterPro family if so specified, not in the user-supplied FASTA file. As in Option B, although the limit on the number of sequences that can be used to generate a SSN is limited to ≤ 150,000, with this advanced option you can select a fraction of the total number of sequences for larger sequence sets to generate a network.
Utility for the identification and coloring of independent clusters within a SSN.

Figure 6. Settings for coloring utility.
The EFI-GNT server for generating genome neighborhood networks (GNNs; http://efi.igb.illinois.edu/efi-gnt/) retrieves genome neighborhood information for sequences in an input SSN. The input SSN is generated by EFI-EST (Options A, B, D, and E; based on UniProt IDs) or exported by Cytoscape after analysis. EFI-GNT recognizes the clusters in the SSN and extracts the UniProt IDs for the sequences in each cluster. Each cluster is assigned a unique cluster number, and the nodes for the sequences in each cluster are assigned a unique color. This “colored SSN” is available for download, along with the GNNs. The colored SSN assists the user in analyzing the GNNs by allowing color-guided association of the cluster nodes in the GNNs with the clusters in the input SSN.
However, a colored SSN also is useful for analyses of SSNs. For example, instead of analyzing a monochromatic SSN, the colored SSN may provide the ability to more easily locate and identify clusters in complicated SSNs.
Also, the colors in a colored SSN can be used to identify how isofunctional clusters emerge as the alignment score is increased (vide infra). Sequences in clusters that are intermingled at low values of the alignment score and segregate into separate clusters as the alignment score is increased may share functional properties. This tracking of cluster separation is made “easy” if the colors assigned to the clusters in the “final” colored SSN with segregated clusters can be assigned to the nodes/sequences in SSNs filtered with smaller alignment scores.
Therefore, we now offer a utility with which the user can upload a SSN with segregated clusters (generated with Option A, B, D, and E or modified with Cytoscape) and, after processing, download a “colored SSN”. The utility also provides:
- A folder with FASTA-formatted files for each cluster in the SSN; these files can be used as the input for other applications, e.g., multiple sequence alignment (MSA) applications such as CLUSTALW.
- A tab-delimited text file that lists each node/sequence in the input SSN with the assigned cluster color and number; this file can be used to add custom node attributes to SSNs filtered with different alignment scores, thereby allowing the user to assess the divergence of clusters as the alignment score increases/percent identity decreases and inferring potential functional relationships.
Click here to contact us for help, reporting issues, or suggestions.
University of Illinois at Urbana-Champaign
1206 W. Gregory Drive Urbana, IL 61801
efi@enzymefunction.org
