
EFI - Chemically-Guided Functional Profiling
"Run CGFP/ShortBRED" tab: EFI-CGFP Input SSNs
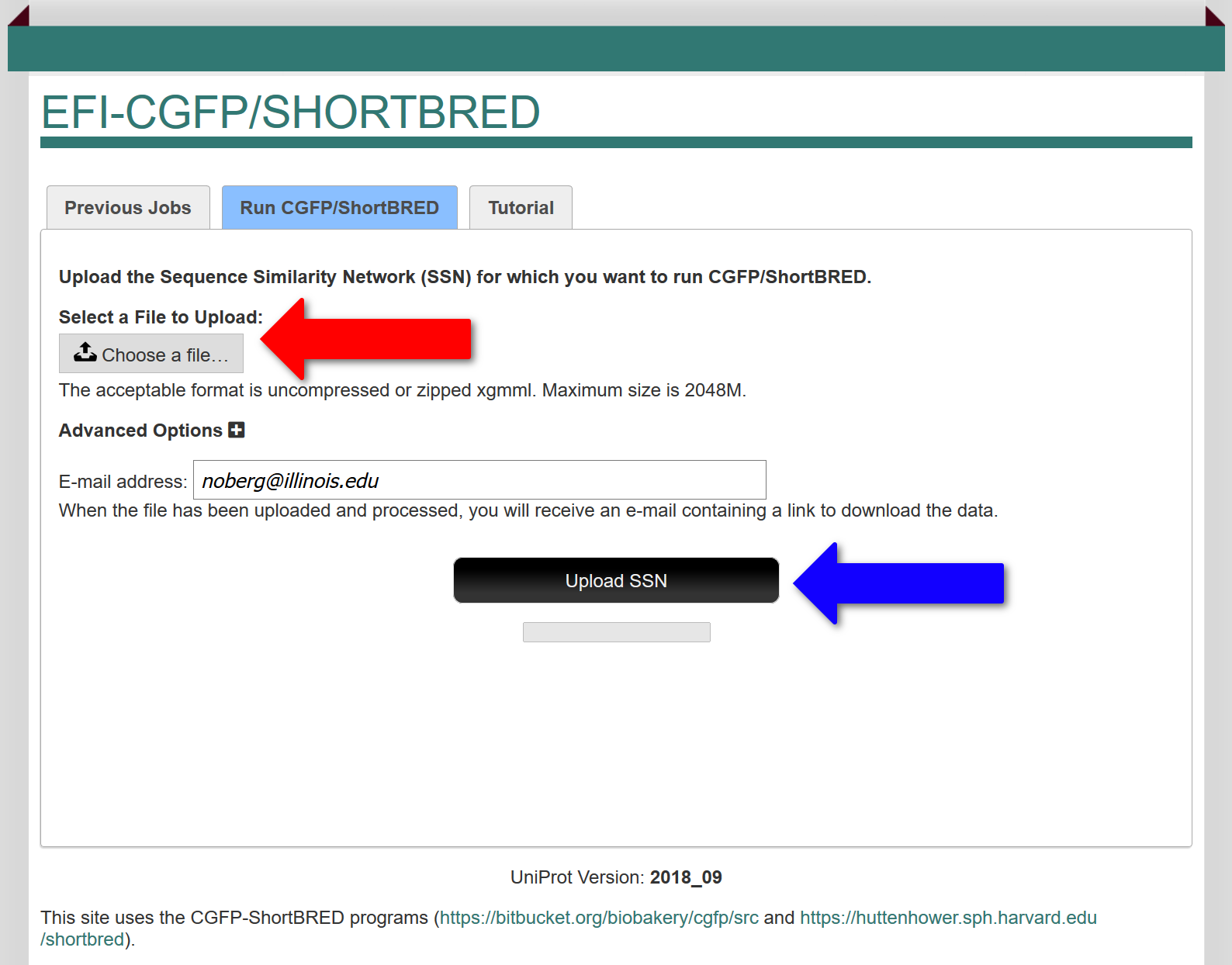
In the "Run CGFP/ShortBRED" tab, the user uploads the colored SSN xgmml file for which the sequence markers are identified (red arrow; either compressed/zipped or uncompressed) and metagenome abundances determined and mapped to the SSN clusters and singletons. Ideally, the input SSN should have been segregated into isofunctional clusters by the user so that EFI-CGFP will allow orthologues of the members of these clusters to be identified in the metagenome datasets. If the user later decides that the SSN clustering can be improved, the user can upload a "child" SSN derived from the initial SSN generated with a different alignment score and/or containing a subset of the sequences/clusters in the original SSN and use the abundances determined with the original SSN to generate heatmaps and abundance statistics for the child SSN.
The input SSN MUST be a Colored SSN generated with either the Color SSN utility of EFI-EST or EFI-GNT. The node attributes for a Colored SSN include "Cluster Number" and "Singleton Number" for each (meta)node [the former if the (meta)node is located in a cluster with ≥2 accession IDs; the latter if the (meta)node is a singleton (a single accession ID)]. These numbers are used to identify the clusters and singletons with metagenome hits both in the input SSN, heat maps, and tables generated by the EFI-CGFP quantify step ("Quantify Results" page, vide infra).
Before they are colored, the SSNs can be of two types: 1) SSNs generated with Options A, B, or D of EFI-EST with sequences derived entirely from the UniProt database (https://www.uniprot.org/) and 2) SSNs generated with Option C of EFI-EST that include sequences provided in a user-provided FASTA file so that they need not be included in the UniProt database, e.g., in the NCBI database or proprietary sequences obtained from in-house sequencing projects. In generating the original SSN, the user may elect to read the FASTA headers (e.g., for sequences from NCBI) to obtain node attributes if an equivalent accession ID can be found in the UniProt database; however, these node attributes are not used by EFI-CGFP.
For SSNs generated with Option C, the sequences provided in the FASTA file are used by EFI-CGFP; the SSNs also may include nodes/sequences for a user-specified UniProt or InterPro family (using the Advanced Options of Option C). EFI-CGFP uses the sequences in the FASTA files and/or those associated with the UniProt IDs in user-specified UniProt and/or InterPro family/families to identify the markers used to interrogate the metagenome datasets in the Quantify step; it does not use any of the node attributes.
The input SSN can be a full SSN with a node for each sequence/accession ID or a representative node (rep node) SSN in which the sequences/accession IDs are grouped in metanodes that share specified levels of sequence identity by EFI-EST. The SSN can be generated using either the complete set of family sequences (for families with ≤25,000 sequences) or UniRef90 seed sequences/clusters (for families with >25,000 sequences, as required by EFI-EST).
For large protein families (>25K sequences), the user may find it useful to generate the input SSN using UniRef50 seed sequences/clusters; this feature is now available on the EFI-EST web tool. The accession IDs in UniRef50 clusters share ≥50% sequence identity over ≥80% of the length of seed sequence. In many cases, UniRef50 clusters are isofunctional. SSNs generated using UniRef50 seed sequences/clusters are "equivalent" to the clustering in 50% rep node SSNs.
SSNs generated with UniRef50 seed sequences/clusters should be generated using alignments scores that correspond to <50% pairwise sequence identity—we recommend alignment scores that correspond to 30-45% pairwise sequence identity. These SSNs will contain fewer (meta)nodes than SSNs generated using UniProt sequences or UniRef90 seed sequences/clusters; the Family Information Page on the EFI-EST tools provides tables that details the number of (seed) sequences for Pfam families/clans and InterPro families for the UniProt, UniRef90, and UniRef50 databases. The node attributes for the SSNs contain the accession IDs that are present in the UniRef90 and UniRef50 clusters, so the user can locate any UniProt accession ID in SSNs generated with the UniRef databases.
Depending on the RAM available on the user's computer, the user may not be able to view a full SSN [complete set of UniProt, UniRef90, or UniRef50 (seed) sequences], but the user should be able to view a rep node SSN. We recommend the highest resolution rep node SSN that can be manipulated with Cytoscape on the user's computer (largest possible sequence identity) so that proteins with different functions can be expected to be located in separate SSN clusters.
We recommend that users apply a minimum length filter to their sequences to ensure that the sequences are "full length" when the input SSN is generated with EFI-EST. The marker identification step in ShortBRED involves initial clustering of proteins into groups of sequences that share a specified level of sequence identity (default is 85%; CD-HIT 85 ShortBRED familes); these sequences then are aligned (multiple sequence alignment using MUSCLE) to generate a consensus sequence. Finally, the consensus sequence is used by the ShortBRED programs to identify unique markers for each CD-HIT 85 ShortBRED family. The presence of sequence "fragments" may bias the multiple sequence alignment/identification of the consensus sequence so they should be avoided/absent in the input SSN.
If the input SSN was generated using UniRef90 or UniRef90 clusters, the minimum/maximum length filters in EFI-EST are applied to the seed sequences for the UniRef clusters (used in the EFI-EST BLAST) and not the sequences in the clusters. [The seed sequence is the longest sequence; shorter sequences that share 90% (UniRef90) or 50% (UniRef50) sequence identity over at least 80% of the length of the seed sequence are located within the cluster.] The user can choose maximum lengths for generation of SSNs, but it should be remembered that some of the sequences in UniRef clusters likely will be shorter than the seed sequence.
The "Run CGFP/ShortBRED" page has an Advanced Option section that provides minimum and maximum length filters that can be applied so that sequences of desired minimum and maximum lengths can be selected from UniRef90 or UniRef50 clusters (see below).
EFI-CGFP identifies and uses only unique sequences (100% sequence identity over 100% of the length of each sequence) in the input SSN so that the consensus sequence is not biased by multiple occurrences of the same sequence; metanodes in rep node SSNs and UniRef90/UniRef50 clusters are expanded so that all sequences included in the metanodes/clusters are used in the identification of unique sequences.
Sequence markers specific to the CD-HIT 85 ShortBRED family consensus sequences are identified by subjecting the consensus sequences to pairwise alignment among themselves and then to pairwise alignment with the sequences in a reference database.
The default parameters for marker identification are 1) those used for clustering the unique sequences into clusters that share 85% sequence identify (CD-HIT 85 ShortBRED families), 2) DIAMOND (normal sensitivity) for the pairwise comparisons of the consensus sequences for the CD-HIT 85 ShortBRED families with one another and a reference sequence database, and 3) the UniRef90 seed sequences for the reference sequence database.
After the quantify (second) step of ShortBRED, the metagenomes identified by the markers for the CD-HIT 85 ShortBRED family seed sequences in an SSN cluster are merged when the cluster abundance is calculated; these are included in a downloadable file as well as summarized in the heatmaps. The output files also include the metagenomes identified by the markers for each of the CD-HIT 85 ShortBRED families.
The Advanced Options section allow the user to change the default parameters used to identify markers for the CD-HIT ShortBRED families:
- The user can select minimum and maximum sequence lengths. As noted previously, UniRef90 and UniRef50 clusters used by EFI-EST to generate SSNs contain sequences shorter than the seed sequence so this option allows minimum and maximum lengths to be specified.
- As an alternative to the UniRef90 seed sequences for the reference sequence dataset, the user can select either the sequences in the full UniProt database or the UniRef50 seed sequences. The UniProt sequences may produce fewer false positive metagenome hits than the default UniRef90 database with a 2-fold longer execution time; UniRef50 may produce more false positive metagenome hits than the default UniRef90 database with a 2-fold shorter execution time.
- The user can specify an alternate sequence identity value for generating the ShortBRED families with CD-HIT, e.g., to ensure that different functions are represented by sequences in different CD-HIT clusters. Proteins that share >60% sequence identity usually have the same functions, but exceptions to this "rule" are known.
- The user can specify BLAST instead of DIAMOND as the algorithm for the pairwise comparisons. BLAST is more "accurate" than DIAMOND but DIAMOND is ~100x faster than BLAST when used by ShortBRED. For large protein families, we recommend the initial use of the DIAMOND default followed, if desired, by more "accurate" analyses of selected SSN clusters using BLAST.
- CGFP retrieves the sequences used for constructing the CD-HIT ShortBRED from the local UniProt database used by EFI-EST. If the SSN was generated with a previous database, that database can be selected to ensure that the sequences will be available (UniProt "retires" sequences so the database used to generate the SSN should be used). The database is given in the Network Information table provided by EFI-EST. The default database is the current database used by EFI-EST.
The quantify step then maps the abundance of metagenome hits to the markers and then to the SSN clusters that contain the CD-HIT ShortBRED families with the markers (next section).
After the colored input SSN is uploaded, marker identification is initiated (with the "Upload SSN" button; blue arrow). The execution time depends on the number of unique sequences. Using DIAMOND the execution time ranges from ~40 minutes for the GRE superfamily (InterPro family IPR004184 with a minimum length filter of 500 residues; ~9000 unique sequences) to ~5 hours for the radical SAM superfamily (InterPro families IPR007197 and IPR006638 without a minimum length filter; ~320K unique sequences). Using BLAST the times are ~24 hrs for the GRE superfamily and three weeks for the radical SAM superfamily.
An e-mail is sent to the user when the input SSN has been uploaded and the marker identification has been initiated. The "Previous Jobs" tab will display the job in black font as soon as it is received and its status will be indicated as "PENDING"; when marker identification has been initiated, the job status will be changed to "RUNNING". When the job is finished, the job name will change to a green-colored link.
Click here to contact us for help, reporting issues, or suggestions.
University of Illinois at Urbana-Champaign
1206 W. Gregory Drive Urbana, IL 61801
efi@enzymefunction.org
